Achieve a 7.4x speedup with 8 GPUs over the performance of a single GPU through the use of task-based parallelism and concurrent kernels! Traditional GPU programming typically views the GPU as a monolithic device that runs a single parallel kernel across the entire device. This approach is fantastic when one kernel can provide enough work to keep the GPU busy. The conundrum is that newer GPUs require ever larger computational problems to keep them fully occupied. The solution is to treat each Streaming Multiprocessor (SMX) inside the GPU as a separate processing core (similar to a single core of a multicore x86 or ARM processor) that runs a separate task list comprised of smaller concurrent kernels. Host-based dynamically scheduled OpenMP loops can then very simply and effectively queue these concurrent task lists as GPU SMX processors become available. This mini-tutorial is the first in a series of techEnablement.com tutorials about GPU task-based parallelism and how to achieve strong-scaling across multiples GPUs as well as the many SMX processors that are internal to each GPU.
The following timeline from nvvp, the NVIDIA Visual Profiler, demonstrates how a single concurrent kernel task-based parallel application can keep multiple GPUs busy in a workstation (in this case an NVIDIA K40 and a K20 GPU) even when the GPUs have widely different performance capabilities. The various colored lines represent different parallel kernels. Note that the length of the lines reprents the runtime of each of the kernels, which can vary in this problem from microseconds to tens of seconds depending on the data input. Overall utilization of both GPUs stays in the 95% – 100% range throughout the application lifespan as reported by the nvidia-smi (NVIDIA Systems Management Interface) tool.

The logic for the load-balancing loop is very simple as shown in the following code snippet. The highlighted pragma performs the load-balancing via schedule(dynamic,1), which does not get blocked by one or more slow threads. The key is to have the task list signal when the task queue starts so more kernels can be asynchronously queued on a CUDA stream by the thread. As a result, all the CUDA stream queues (generally one per SMX) are kept fully loaded with kernels waiting to run.
|
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 |
double startTime=omp_get_wtime(); #pragma omp parallel for schedule(dynamic,1) for(int i=0; i < bootstrap.get_n_proteins(); i++) { int tid=omp_get_thread_num(); bootstrap.calcBootstrap(worker[tid], tid, i ); } for(int i=0; i < worker.size(); i++) { cudaStream_t s = worker[i].setStream(); cudaStreamSynchronize(s); } |
The initial code was developed and tested on an HP Z800 workstation containing three GPUs: A Tesla K40C, A Tesla K20C, and a GeForce GT 640.
The 8-way GPU results showing a 7.4x speedup with 8 GPUs were observed on a Cirrascale BladeRack 2 XL Blade Server configured as if there were 8 GPUs in a single node (e.g. no MPI were involved, only CUDA calls).

Click to learn more

Click on image for more information
Application
The scaling results and timeline are from a statistical bootstrapping code written by TechEnablement.com and BlackDog Endeavors, LLC in collaboration with Dr. John Wilson for the Cold Spring Harbor Scientific Laboratory. This production code will be incorporated into an online interactive booststrapping workflow that will be generally accessible via the Internet in the near future.
A quick synopsis of this work was presented in my GTC 2014 talk “S4178: Killer-app Fundamentals: Massively-parallel data structures, Performance to 13 PF/s, Portability, Transparency, and more ” [pdf][video]. Following are the two pertinent slides
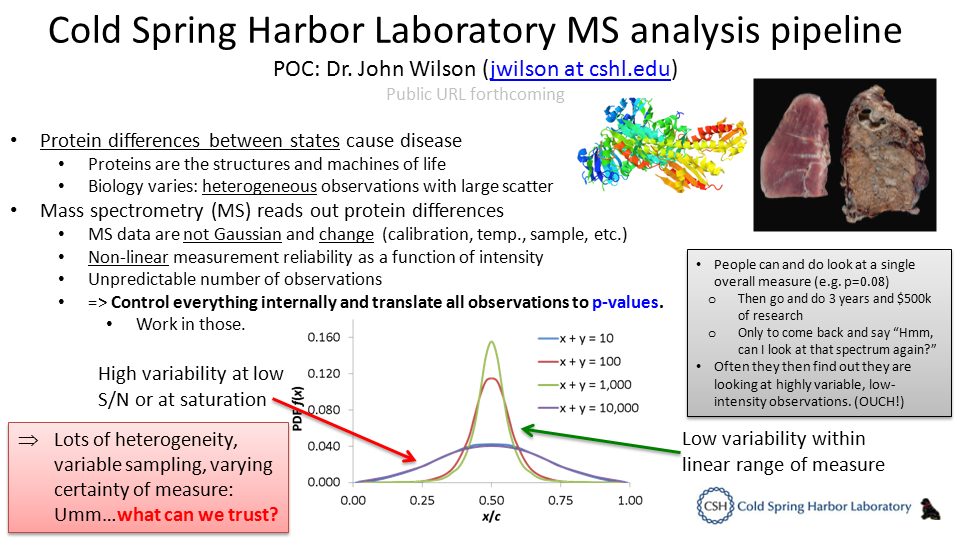

For more information please see the ASMS 2014 poster “A GPU-powered, massively parallel nonparametric statistics server for analysis and exploration of large-scale quantitative data between and across quantitative experiments” by John P. Wilson, Robert M. Farber, and Darryl J.C. Pappin
Wilson et al ASMS2014-statistics-poster final
Following is the poster introduction:
We often employ proteomics in the quest to understand two or more states: what makes this healthy and that diseased? Quantitative techniques such as iTRAQ and SILAC are powerful tools in this pursuit. However, their results immediately beg the question “What’s different?”, making determination of “different” between states essential to distilling meaning from quantitative MS data. Arguably, the most helpful output is some measure of certainty that the variable of study actually caused the difference observed. We have developed a novel experimental and analytical methodology which employs exclusively nonparametric statistics to determine p-values and fold-changes (including their uncertainties with confidence intervals) for observed differences as a function of biological variation, number of samples and observations, and measurement error (most crucially accounting for increased variance at low or high MS/MS spectral intensities). Our workflow uses no parametric modeling nor parametric simplifications, accepts data from all MS quantification techniques, can combine across experiments with ≥1 common control channel and is accessible through an interactive, real-time web interface.
Leave a Reply